آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Chromosomal abnormalities |


|
|
Read More
Date: 11-11-2015
Date: 10-11-2015
Date: 10-11-2015
|
Chromosomal abnormalities
Chromosome abnormalities are changes resulting in a visible alteration of chromosomes. An alternative definition of a chromosomal abnormality is an abnormality produced by specific chromosomal mechanisms. Most aberrations are produced by misrepair of broken chromosomes, improper recombination or improper segregation of chromosomes during mitosis or meiosis. Chromosome abnormalities are an important cause of mortality and morbidity and nearly 50 to 60% of foetal wastage.
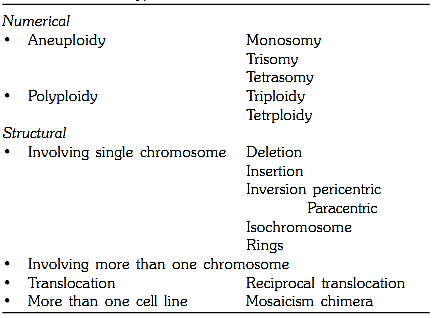
A chromosomal abnormality may be present in all cells of the body (constitutional abnormality) or may be present only in certain cells or tissues (somatic abnormality). Chromosomal abnormalities, whether constitutional or somatic, fall into two categories, numerical and structural abnormalities (Table 1).
Various types of abnormal chromosomal patterns and rearrangements result into classical and non-classical syndromes. These are described in the chapter on chromosomal syndromes. The following pages describe the types of chromosomal abnormalities.
Table 1: Types of chromosomal abnormalities
Numerical chromosomal abnormalities
Numerical abnormalities occur when the normal human chromosomal complement of 46 gets addition or loss of one or more chromosomes in the diploid number (2N). This is termed as aneuploidy. If a chromosomal complement has multiples of haploid number (1N) it is termed as polyploidy.
polyploidy
Cell lines that contain multiples of the haploid number other than diploid are called polyploid. Triploidy (3n) 69 and tetraploidy (4n) 92 (Fig. 1) are the two most commonly seen forms of polyploidy. Triploidy may be due to failure of the ovum or the sperm to divide at maturation. This may also be the result of fertilization of the ovum by two sperm, or fertilization of an ovum that has not expelled the first polar body. The phenotypic expression varies with the source of the extra set of chromosomes. When this extra set is of paternal origin, the foetuses have an abnormal placenta and are classified as hydatidiform moles. Those with an extra complement of maternal origin are aborted spontaneously.
Three sex types have been observed in triploidy, these being 69,XXX, 69,XXY and 69,XYY Triploidy can result in abortions or in some cases live births that die at or shortly after birth. Tetraploidy may be seen as an artefact of tissue culture. True tetraploidy is very rare. All live born non-mosaics of polyploidy have died within a few hours of birth. Tetraploids are usually
92,XXXX or 92,XXYY. This is an indication that the cause of tetraploidy is the failure of completion of an early cleavage division of the zygote. Endoreduplication arises from failure of the centromeres to separate during anaphase; and this may be observed in tissue culture. The chromatids undergo the DNA synthesis phase for a second time and appear as four chromatids fastened at the centromere. This is a rare occurrence in vivo. Tumour cells may show a polyploid complement as a result of endoreduplication.

Figs 1A and B: Metaphase spreads showing (A) Teraploidy and (B) Triploidy
Aneuploidy
When a single chromosome is added to the normal chromosomal complement it is called trisomy. When two chromosomes are added it is called tetrasomy. When there is a loss of a single chromosome from the normal chromosomal complement it is called monosomy.
Numerous chromosomal abnormalities involving the loss or gain of an entire chromosome have been reported, many being seen only in spontaneously aborted foetuses. These are briefly mentioned below and discussed in chapter on Chromosomal disorders.
Trisomy
However, there are three well-defined chromosomal disorders that are compatible with postnatal survival. The 3 well recognized trisomies for an autosome are trisomy 21 (Down syndrome), trisomy 18 (Edward syndrome), and trisomy 13 (Fig. 2) (Patau syndrome). Each of these autosomal trisomies is seen to be associated with growth retardation, mental retardation and multiple systemic anomalies. Though each has a distinctive phenotype, there can be variation in expression or in severity and involvement of systems. Trisomies other than these usually result in pregnancy loss. Trisomy 16 is a common trisomy of the autosome seen in first trimester foetal losses. The commonly known syndromes are 47, XXY (Klinefelter syndrome), and less commonly 47, XYY. Multiples of X chromosome syndromes are known and have same phenotypic effect in males. In females 47, XXX Triple X syndrome is known. Presence of an extra sex chromosome is mostly compatible with life and has very few phenotypic effects.


Figs 2 A to C: Metaphase spreads showing (A) trisomy 21, (B) trisomy 18 and (C) trisomy 13
Monosomy
The term monosomy is absence of a single chromosome from a normal diploid complement. Autosomal monosomies are always lethal. Sex chromosomal monosomies are compatible with life but could also result in foetal loss. The most common example is 45,X (Turner syndrome). Turner syndrome can be due to loss of either an X or Y chromosome. The cause of monosomy is non-disjunction at meiosis. If during divisions one gamete receives two copies of a homologous pair, the other gamete will have absence of a chromosome in it (nullisomy). Monosomies are also known to occur due to no anaphase lag, leading to loss of chromosomes.
Origin of Trisomies and Monosomies
Trisomies mainly occur by failure of separation of homologues chromosomes at meiosis I (anaphase) (Fig. 3). This is called non-disjunction. Trisomies can also occur due to non-disjunction at meiosis II. Here the sister chromatids of a pair fail to dissociate. In both the cases, the gamete gets a pair of homologous chromosomes. At fertilization, a single chromosome from a parent results in trisomy. Most trisomies occur due to non-disjunction at maternal meiosis. Another group of trisomies resulting from non-disjunction occur in a developing zygote during early mitotic divisions. Such divisions usually lead to mosaic cell lines.
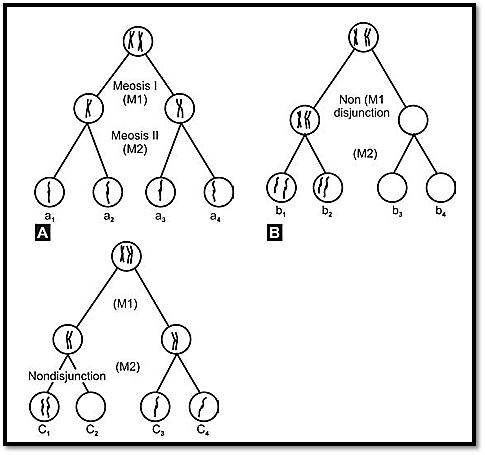
Fig. 3: Mechanisms of non-dysjunction producing disomic and nullisomic gametes) (A) normal (B) M1-nondysjunction (C) M2-nondysjunction
Causes of Nondisjunction
We have seen that nondisjunction leads to numerical errors of chromosomes. But what causes nondisjunction is still uncertain. Increased incidence of Down’s syndrome in advanced maternal age suggests an effect of aging on the primary oocytes. Trisomy 13 and 18 can also occur with advancing maternal age. In a female the primary oocyte lies in suspended prophase stage. This means an egg of a female is as old as she is. The theory put forward for maternal age and disjunction is that there may be absence of recombination between homologous chromosomes in the ovary of the foetus. Incidence of aneuploidy is also increased when there is delay between ovulation and fertilization.
Structural chromosomal abnormalities
Structural rearrangements are a result of chromosome breakage and reunion at an abnormal site. Such abnormalities are usually heritable and are a cause for chromosomal aberrations in the progeny. Cells have enzymes for repair of broken strands of DNA and such repair goes on throughout the life of each cell. Some preference sites for breaks are known and are called fragile sites. Chromosome breakage is frequently accompanied by exchange of material from one chromatid to another during mitosis, when the replicated chromosomes are waiting to separate into two daughter cells. This is known as sister chromatid exchange (SCE). During meiosis, exchange of material on homologous chromosomes occurs during pachytene of the first meiotic division. This ensures mixing of the maternal and paternal gene pool and is termed crossing over. SCE and crossing over are seen in somatic and germ cells respectively. Abnormalities arise only if the chromosomes break at non- homologous sites leading to unequal exchanges.
Rearrangements can occur within a chromosome or may involve more than one chromosome. An individual with a normal chromosomal set is said to have balanced chromosomes. If some information is additional or missing, the arrangement is called an unbalanced chromosomal arrangement. Balanced rearrangements normally do not cause any phenotypic effect, as all the genetic information is present even though at a different position. The subsequent generations however, are at a risk, as such carriers are likely to produce unbalanced gametes resulting in abnormal offspring with unbalanced karyotypes.
Rearrangements involving single chromosomes
The phenotype is likely to be abnormal because of deletion, duplication, or in some cases, both. Duplication of a part of a chromosome is comparable with partial trisomy; deletion leads to partial monosomy. Any change that leads to deviation from the normal genetic complement may result in abnormal development.
Deletion
Deletion is a loss of chromosomal material causing an imbalance in the normal complement. The clinical manifestations depend on the size of the deleted portion and the function of the genes in that segment. Deletion may occur due to chromosome breakage within the chromosome. If the pieces are reconnected to the acentric material, the resultant chromosome is short (Fig. 4). Deletions may also be generated by abnormal segregation from a balanced translocation or inversion. This appears to be a more likely mechanism than multiple breaks in a single chromosome. A deletion may be terminal or interstitial. High-resolution banding may be used in cases of deletions that are not observable by routine metaphase studies. To be detectable by high-resolution banding, a deletion must be at least 2-3 megabases in size. FISH techniques may be
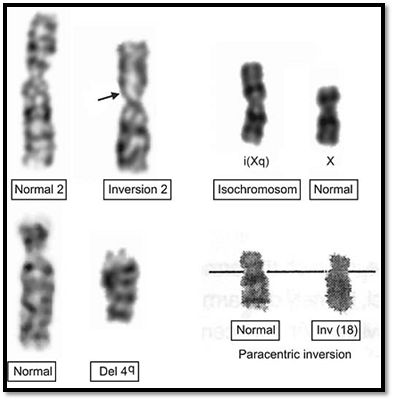
Fig. 4: Examples of chromosomal anomalies involving a single chromosome
used for detection of very small deletions. Specific syndromes have been ascribed to certain deletions and are described in the section on microdeletion syndromes, in the chapter on chromosomal syndromes. Deletions that appear to be identical in extent but different in parental origin may lead to differences in phenotypic expression. This is due to genomic imprinting, which marks maternal and paternal chromosomes differently. An example of this is the Prader-Willi and Angelman syndromes.
Duplication
A duplicated segment may be inserted in the same order as the original segment or may be reversed. Tandem duplications may arise by unequal crossing over during meiosis or from a rearrangement between two chromatids during mitosis. To form a reversed duplication, the segment should be inserted upside down next to the original segment. The exact mechanism of this rearrangement is not known. Duplication is usually less harmful than a deletion. However, because duplication in a gamete results in chromosomal imbalance, and because of the chromosome breaks that generate, it may disrupt genes. Duplication often leads to some sort of a phenotypic abnormality. Certain phenotypes appear to be associated with duplications of particular chromosomal regions and are functionally trisomic for the regions.
Inversion
An inversion involves two breaks in a single chromosome. The broken segment turns a complete 180° and reattaches to the points of breaks. Two types of inversions are known, paracentric or pericentric (Fig. 4). The centromere is not included in a paracentric inversion as both breaks occur in one arm, hence the arm ratio is unchanged. In a pericentric inversion the centromere is included in the inverted portion, causing the arm ratio to change. As no change is involved in the arm ratio in paracentric inversions, they can be detected by banded preparations. Pericentric inversions are easier to identify as the arm ratio and the banding pattern is altered (Fig. 5).
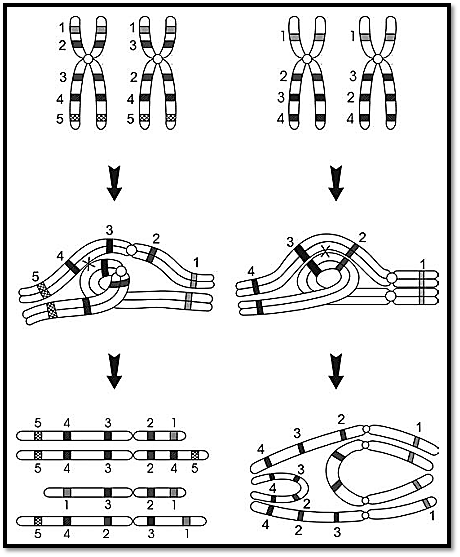
Fig. 5: Pericentric and paracentric inversions and mechanisms of production of recombinant chromosomes
An inversion does not usually cause any phenotypic change, as it is a type of balanced rearrangement. A carrier of either type of inversion is at a risk for producing abnormal gametes that may lead to unbalanced chromosomal complements in the offspring. The manifestation of the two types of inversions is different. A loop is formed when the chromosomes with an inversion, pair in meiosis I; if crossing over occurs within the loop, a deleted or duplicated chromosome can result (Fig. 5). Inversions are only rarely implicated in chromosomal abnormalities in humans. Recombination which is a normal feature of meiosis I, is somewhat suppressed within inversion loops, but may occur in larger inversions. When the inversion is paracentric, acentric or dicentric chromosomes are formed on recombination and the resulting gametes with an unbalanced complement may not be compatible with the survival of the offspring. A pericentric inversion may result in unbalanced gametes with duplication or a deficiency of chromosome segments flanking the site of inversion. A particular risk is associated with pericentric inversions; the larger ones being more likely to result in viable offspring than smaller ones, because the former have smaller unbalanced segments. Pericentric inversion in chromosome 9 is the most commonly seen chromosomal inversion in humans. An increased risk of miscarriage is not commonly seen and these are considered normal variants, as there does not appear to be an increased risk of producing unbalanced gametes.
Isochromosomes
An isochromosome is one in which the arms on either side of the centromere are morphologically identical and bear the same genetic loci, namely one arm is missing while the other is reduplicated. Isochromosomes may be formed by horizontal division of the centromere instead of vertical division. Thus the two arms of the chromosome are separated instead of two chromatids. In subsequent mitosis the joint arms act as a bi-armed chromosome. Formation of isochromosomes may occur by chromatid-to-chromatid exchange, or chromatid translocations within a chromosome following breakage and loss of the distal sections of the chromatids. This may cause many isochromosomes to be dicentric. Isochromosomes appearing monocentric may have two centromeres so close to each other that they cannot be perceived as separate; special staining may be required to visualize them.
The isochromosome of the long arm of the X chromosome, denoted as i (Xq) is the most commonly seen isochromosome, observed in some individuals with Turner syndrome. Isochromosome 17q is seen in some patients with leukaemia. Solid tumours may also show isochromosomes . Isochromosomes have also been seen in chromosome 12, 13, 18 and 21. The clinical effects manifested by isochromosomes are a result of the monosomic state of the missing loci, as well as the trisomic state of the genes on the isochromosome.
Ring Chromosomes
Ring chromosomes are a result of the joining together of the sticky ends caused by two breaks in a single chromosome (Fig. 6). The two terminal fragments are lost, giving rise to monosomic state of these loci. Clinical manifestations are a result of monosomy. If the centromere is within the ring, fragments lost are acentric. Disjoining of ring chromosomes at anaphase may pose a problem, especially when a twist is developed in a ring through breakage and reunion. Breakage and fusion may form larger and smaller rings. Because of mitotic instability, ring chromosomes may be seen only in a proportion of cells. Ring chromosomes have been detected for every human chromosome. Presence of a ring of any type can lead to ring syndrome, because of random duplication and deletion of genetic material in many different cell lines.
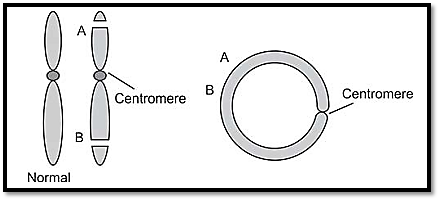
Fig. 6: Mechanism of formation of ring chromosomes
Dicentric Chromosomes
A dicentric chromosome possesses two centromeres, resulting from the joining of two broken fragments of chromosomes, each having a centromere. These may be formed from two different chromosomes or from two chromatids of the same chromosome. The two centromeres may act as a single large one if they are situated very near each other, or one may be inactivated in this case (sometimes called ‘pseudodicentric’). If the centromeres are far apart or if both are active, they can be drawn to opposite poles of the spindle, resulting in formation of an anaphase bridge, a chromosome that makes a bridge between two daughter cells at anaphase. This may result in the dicentrics being left outside both the daughter nuclei as they form, or in breaking apart, leading to a loss or gain of chromosomal material. Dicentric chromosomes are most likely to be observed in cancer cells and represent an acquired abnormality. The most common dicentrics and pseudodicentrics are formed from the acrocentric D and G group chromosomes. Other chromosomes might be involved occasionally.
Rearrangements involving more than one chromosome
Translocations
Translocation involves exchange of genetic material between two or more non-homologous chromosomes. This can occur when two or more chromosomes break at the same time. Broken ends are usually sticky and the cellular enzymatic repair service usually reunites them, but occasionally a mismatch is possible. Breakage tends to occur more frequently at fragile sites at or near the centromere, at chromosome ends or at euchromatin- heterochromatin junctions.
Translocations are classified as reciprocal translocations or Robertsonian translocation.
Reciprocal Translocations
This type of rearrangement occurs when the breakage of non- homologous chromosomes results in reciprocal exchange of the broken segments. Usually only two chromosomes are involved. As the exchange is reciprocal, the total chromosome number is unchanged. In very rare situations three or more chromosomes may be involved. Reciprocal translocations are usually harmless as they are balanced rearrangements. However, they have a risk of producing unbalanced gametes and abnormal progeny. There may be meiotic complications, particularly a risk of non-disjunction.
Robertsonian Translocations
In this type of translocation two acrocentric chromosomes fuse near the centromere region with loss of the short arms. The resulting balanced karyotype has only 45 chromosomes, one of them consisting of the long arms of two chromosomes (Fig. 7B). Because the short arms of the acrocentric
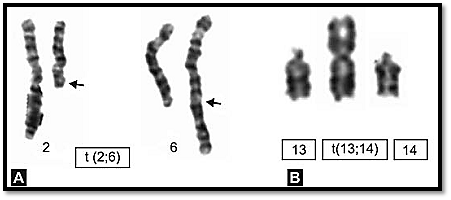
chromosomes have multiple copies of genes for ribosomal RNA, the loss of their short arms is not deleterious. Phenotypically, Robertsonian translocation carriers may be normal but there is an increased risk of production of unbalanced gametes and therefore of abnormal offspring. Of clinical importance is the one involving chromosome 21 as there is a risk of producing a child with translocation Down’s syndrome. It is difficult to decide which centromere of a chromosome is involved unless C or Q banding is done. The numerical count in Robertsonian translocation is 45.
A translocation of either type can render the carrier functionally sterile, because of the complex synaptic structures formed. Complex translocations involving more than two breaks can cause serious problems in cell division. Small exchanges of the genetic material may produce viable dysmorphic infants, whereas large exchanges may lead to greater problems with spontaneous abortions. Sporadic translocation in chromosome 7;14 occurs in PHA stimulated blood samples.
Insertions
These are non-reciprocal type of translocations as a segment removed from one chromosome is inserted into a different chromosome. This insertion is either in its usual orientation or in an inverted one. Insertions are however, rare, as they require three breaks. Abnormal segregation in an insertion carrier can produce offspring with duplication or deletion of the inserted segment, as well as normal offspring and balanced carriers.
Marker chromosomes
Marker chromosomes are occasionally seen in tissue culture, mostly in the mosaic state. They are designated as supernumerary chromosomes, as they are present in addition to the normal chromosomal complement. A marker chromosome also comprises a structural rearrangement. A marker chromosome must have a centromere. It may be derived from breakage of a chromosome with loss of the acentric fragment and non-disjunction from its homologue at meiosis. Tiny markers often consist of little more than centric heterochromatin, whereas larger ones contain some material from one or both arms, creating an imbalance for whatever genes are present. Due to problems in identification of the marker chromosomes, its clinical significance is difficult to assess, and hence poses serious problems in genetic counselling. In some cases, no phenotypic effects have been seen in individuals with small markers. Some others however produce severe clinical effects. If a marker chromosome has an identifiable centromere, it should be included as a derivative chromosome (der); if no further identification is possible, it should be denoted by the marker symbol (mar). If a marker chromosome is observed in amniotic fluid culture or chorionic villous samples, a prenatal karyotype is recommended to confirm its origin as familial or de novo.
Breaks and gaps
Breaks can occur in chromatids or in chromosomes. In a break the chromosomal segment is completely fractured. The separate segment is either lost, or is seen as an attached fragment. In a gap, the segment appears discontinuous, but is attached by a thread like structure. Chromosomal breaks and gaps are significant, as they involve a loss of chromosomal material.
Genetic imprinting and uniparental disomy
An individual inheriting two copies of the same homologous gene from one parent due to an error at meiosis II, is an example of uniparental disomy. An individual inheriting two different homologues from any one parent and through error at meiosis I will have uniparental hetero-disomy. In both the above conditions, the conceptus would be trisomic, with loss of a chromosome resulting in a disomic State. One third of foetuses with such chromosome losses would result in uniparental disomy.
Genomic imprinting is defined as determination of the expression of a gene by its parental origin. It is generally accepted that an individual inherits one autosomal allele from each parent and that these alleles are equally expressed. Exceptions to this rule were detected for two syndromes Prader-Willi (PWS) and Angelman syndrome (AS). Both these syndromes are caused in most instances by microdeletions of the same chromosomal region on 15q11-q13. However, in PWS, the individuals inherit the deleted chromosome from their father and in AS the individuals inherit their deleted chromosome from their mother. This is thought to be due to functional inactivation (imprinting) of the nondeleted homologue (Fig. 8), resulting in structural monosomy, but functional nullisomy. Whereas maternal uniparental disomy resulting in PWS is common, accounting for about 20% of all cases, disomy of the paternal chromosome 15causing as is rarer.
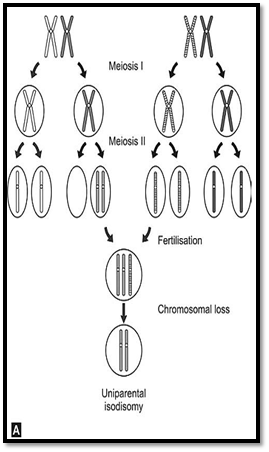
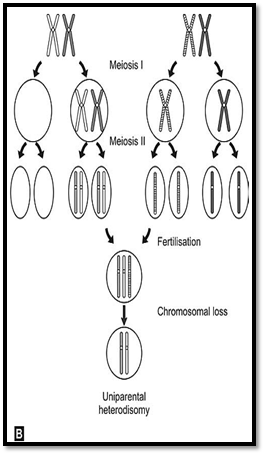
Figs 8A and B: Mechanism of origin of uniparental disomy (A) Uniparental isodisomy (B) Uniparental heterodisomy
Other chromosomal abnormalities
Mosaicism
The term mosaicism is applied to a condition where a in a body tissue, more than one cell type or line is seen. This can occur at mitosis, or any time after conception. Mosaicism can be present at two levels, in somatic cells or in gonadal cells (germ cells).
Somatic Mosaicism
When the phenotype of a single gene disorder is less severe in an individual or is confined to a specific body part, somatic mosaicism should be suspected. The mutation pattern and severity will depend on the time when it arises during the developmental process. Hypomelanosis of ITO (Fig. 9) a disorder showing alternating patterns of pigmented and depigmented streaks corresponding to embryological developmental lines (Blase hos lines).
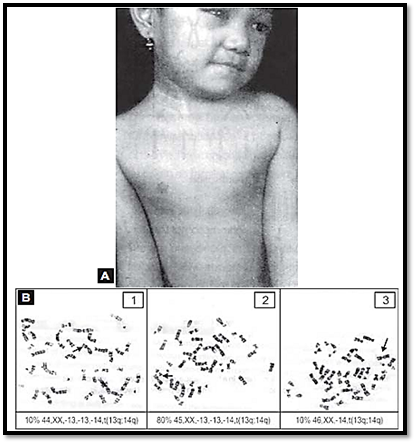
Fig. 9: Patterns of skin pigmentation on the body of a female child having mosaic cell line Metaphase spreads below.The skin conditions is called hypomelanosis of ITO ( Figure 3.20A for color version see Plate 2)
Gonadal Mosaicism
There are certain families where a known genetic inheritance pattern like autosomal dominant or recessive is inherited, and more than one child is affected in spite of the parents being normal. This can be explained by gonadal mosaicism, where the mutation occurs only in the parents germ-line, and therefore the parents are not affected and are normal.
By definition, mosaicism is the presence of two or more chromosomally distinct cell lines. This may arise due to nondisjunction during early division of the zygote, or due to anaphase lag. In anaphase lag, there is a delay in chromosome movement on the spindle, and it does not reach the daughter cell before the nuclear membrane closes. In such a type of mosaicism, this is transmitted as an abnormal cell line, but the other cells of the embryo are normal.
If during chromosomal counting a mosaic cell line is observed, additional cells (a total of up to 100) should be counted, to ascertain the percentage of mosaicism. The level of mosaicism depends on when the misdivision occurs. If it is at the first cell division after fertilization, most of the body tissues will be affected. If the misdivision occurs after the formation of three germ layers ectoderm, mesoderm and endoderm, the abnormal cell line may be present in only one cell type.
Mosaicism can also be acquired. This is noted in cytogenetic analysis of malignant tissues or because of the impact of viruses and chemicals. It is recommended that 20 cells be counted and if they have an equal modal number, it is sufficient to give a diagnosis. If at any time, clinical diagnosis suggests the presence of a syndrome, additional cells are to be counted. In case of cancer patients or for investigating fragile site or chromosomal instability syndromes it is necessary to analyse more cells. If a single cell with a different modal number is found in the usual counting of 20 cells, an additional counting of up to 30 cells is indicated. If no further cells are noted mosaicism can be assumed to be an artefact.
In case of prenatal diagnostic samples, mosaicism for chromosome number 2 and 16 is observed as a common artefact. With bone marrow samples even if a single aberrant cell is observed, it needs to be reported, as it can be significant. The term pseudomosaicism is used for sporadic artifactual changes. It is important to differentiate between the two.
Chimera
Another condition of a mosaic cell line is known as chimera. A chimera has cells of different genetic constitutions. Here, two cell lines originate from two separate zygotes by fertilization of a polar body and the ovum. These two subsequently fuse. Chimeras can also arise by fertilization of two ova, which then fuse for example an XX / XY cell line.
Hydatidiform Moles
Paternally derived genes are responsible and essential for trophoblast development and maternally derived for early embryonic development. In hydatidiform moles, the pathology lies in the placental tissue. The placental morphology is completely distorted. Hydatidiform mole can be classified as a partial or a complete mole. In a partial mole, the foetus is always present but it rarely survives to term. In partial moles the conceptus is always triploid. Using DNA polymorphism studies it has been shown that the father contributes 46 chromosomes. This diploid paternal chromosome is either due to fertilization by two sperm or dispermy or duplication of haploid sperm chromosomes by endoreduplication.
Complete Hydatidiform Mole
Complete moles have 46 chromosomes exclusively o paternal origin. The condition is caused by fertilization of an empty ovum by two sperms or endoreduplication of a single sperm as in a partial mole. Complete moles are of importance in obstetric management as they are liable to undergo malignant change into invasive choriocarcinoma. Successful management of choriocarcinoma is possible by chemotherapy, but in untreated patients the outcome is fatal.
Karyotype Reporting
There is a refined system for reporting a karyotype (Table 2). The first point is to give total number of chromosomes including sex chromosomes, followed by a comma (,), the sex chromosomes are given next. If there is an abnormality of autosomes, that is specified next. Thus, a normal female karyotype is reported as 46,XX normal female and that of a normal male as 46,XY normal male.
If there is sex chromosomal aberration, it is written first. In addition, if autosomal abnormalities are noted they are written next in numerical order e.g. 47,X, t (X; 13) (q27; q12) In uncomplicated cases a karyotype is written as follows:
- First the total number of chromosomes is written, then the sex chromosomes and next the addition of any chromosome if present.
- 45,X - (loss of one ‘X’ chromosome as in Turner’s Syndrome)
- 47,XX, +21 (for Down’s Syndrome)
In a mosaic cell-line, both the cell lines are separated by a slash.
- 45,X/46,XY In case of mosaic cell line the major cell line is described first and the number of cell counted is given in the following square brackets 45,X [27]/46,XY [23]. In addition, symbols are used in rearrangements. The symbol is placed ahead of the chromosome involved and the involved chromosome is written in the parenthesis. 46,XX, r(20), means a female karyotype with a ring form of chromosome 20.
Table 2: Common nomenclature symbols and abbreviation

For Banded Chromosomes
Regions and bands are numbered from the centromere. The symbol p is designated to the short arm and q to the long arm of the chromosome. The centromere is designated as 10. The part adjacent it on the short arm is p10 and on the long arm, q10. The regions adjacent to the centromere are labelled 1 on both the arms, the distal regions as 2 and so on. The band designation is written as follows:
1- Chromosome number
2- Arm symbol
3- Region number
4- Band number within the region.
There is no spacing or punctuation. For example, 1p31 indicates chromosome 1, short arm region 3, band 1, if the band has a subdivision, a decimal point is placed after the band description e.g. 1p3 is subdivided further in to three subbands 1p31.1, 1p31.2 and 1p31.3.
References
Purandarey , H. (2009) . Essentials of Human Genetics. Second Edition. Jaypee Brothers Medical Publishers (P) Ltd.

|
|
|
|
لشعر لامع وكثيف وصحي.. وصفة تكشف "سرا آسيويا" قديما
|
|
|
|
|
|
|
كيفية الحفاظ على فرامل السيارة لضمان الأمان المثالي
|
|
|
|
|
|
|
العتبة العباسية المقدسة تجري القرعة الخاصة بأداء مناسك الحج لمنتسبيها
|
|
|