علم الكيمياء

تاريخ الكيمياء والعلماء المشاهير
التحاضير والتجارب الكيميائية
المخاطر والوقاية في الكيمياء
اخرى
مقالات متنوعة في علم الكيمياء
كيمياء عامة
الكيمياء التحليلية
مواضيع عامة في الكيمياء التحليلية
التحليل النوعي والكمي
التحليل الآلي (الطيفي)
طرق الفصل والتنقية
الكيمياء الحياتية
مواضيع عامة في الكيمياء الحياتية
الكاربوهيدرات
الاحماض الامينية والبروتينات
الانزيمات
الدهون
الاحماض النووية
الفيتامينات والمرافقات الانزيمية
الهرمونات
الكيمياء العضوية
مواضيع عامة في الكيمياء العضوية
الهايدروكاربونات
المركبات الوسطية وميكانيكيات التفاعلات العضوية
التشخيص العضوي
تجارب وتفاعلات في الكيمياء العضوية
الكيمياء الفيزيائية
مواضيع عامة في الكيمياء الفيزيائية
الكيمياء الحرارية
حركية التفاعلات الكيميائية
الكيمياء الكهربائية
الكيمياء اللاعضوية
مواضيع عامة في الكيمياء اللاعضوية
الجدول الدوري وخواص العناصر
نظريات التآصر الكيميائي
كيمياء العناصر الانتقالية ومركباتها المعقدة
مواضيع اخرى في الكيمياء
كيمياء النانو
الكيمياء السريرية
الكيمياء الطبية والدوائية
كيمياء الاغذية والنواتج الطبيعية
الكيمياء الجنائية
الكيمياء الصناعية
البترو كيمياويات
الكيمياء الخضراء
كيمياء البيئة
كيمياء البوليمرات
مواضيع عامة في الكيمياء الصناعية
الكيمياء التناسقية
الكيمياء الاشعاعية والنووية

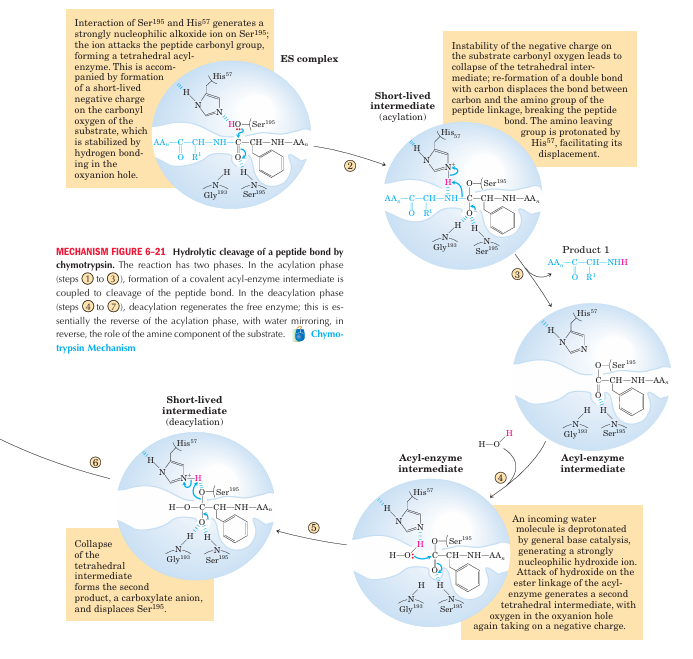
Examples of Enzymatic Reactions:-The Chymotrypsin Mechanism Involves Acylation and Deacylation of a Ser Residue
 المؤلف:
David L. Nelson، Michael M. Cox
المؤلف:
David L. Nelson، Michael M. Cox
 المصدر:
Lehninger Principles of Biochemistry
المصدر:
Lehninger Principles of Biochemistry
 الجزء والصفحة:
P213-218
الجزء والصفحة:
P213-218
 2026-04-26
2026-04-26
 57
57




+
-
20
Examples of Enzymatic Reactions:-The Chymotrypsin Mechanism Involves Acylation and Deacylation of a Ser Residue
Bovine pancreatic chymotrypsin (Mr 25,191) is a pro tease, an enzyme that catalyzes the hydrolytic cleavage of peptide bonds. This protease is specific for peptide bonds adjacent to aromatic amino acid residues (Trp, Phe, Tyr). The three-dimensional structure of chymotrypsin is shown in Figure 6–18, with functional groups in the active site emphasized. The reaction catalyzed by this enzyme illustrates the principle of transition-state stabilization and also provides a classic example of general acid-base catalysis and covalent catalysis. Chymotrypsin enhances the rate of peptide bond hydrolysis by a factor of at least 109. It does not catalyze a direct attack of water on the peptide bond; instead, a transient covalent acyl-enzyme intermediate is formed. The reaction thus has two distinct phases. In the acylation phase, the peptide bond is cleaved and an ester linkage is formed between the peptide carbonyl carbon and the enzyme. In the deacylation phase, the ester linkage is hydrolyzed and the nonacylated enzyme regenerated.
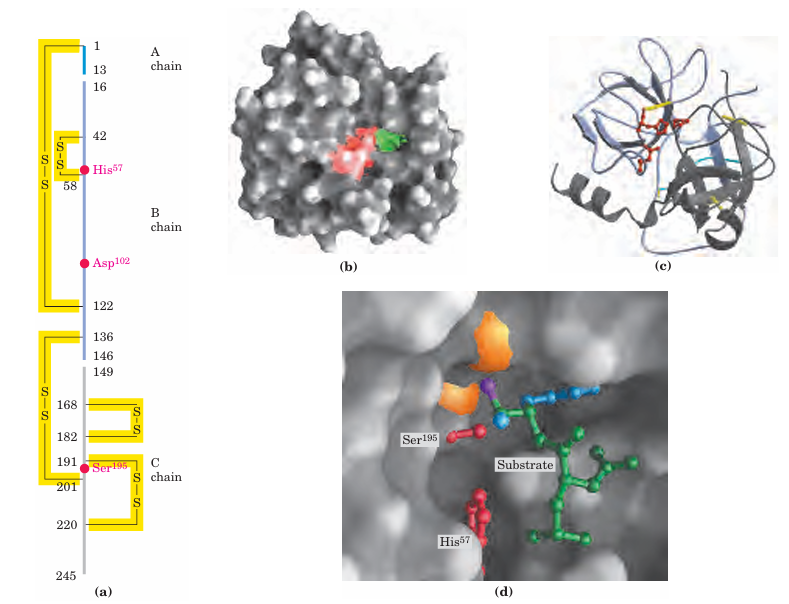
FIGURE 6–18 Structure of chymotrypsin. (PDB ID 7GCH) (a) A representation of primary structure, showing disulfide bonds and the amino acid residues crucial to catalysis. The protein consists of three polypeptide chains linked by disulfide bonds. (The numbering of residues in chymotrypsin, with “missing” residues 14, 15, 147, and 148, is explained in Fig. 6–33.) The active-site amino acid residues are grouped together in the three-dimensional structure. (b) A depiction of the enzyme emphasizing its surface. The pocket in which the aromatic amino acid side chain of the substrate is bound is shown in green. Key active-site residues, including Ser195, His57, and Asp102, are red. The roles of these residues in catalysis are illustrated in Fig ure 6–21. (c) The polypeptide backbone as a ribbon structure. Disul fide bonds are yellow; the three chains are colored as in part (a). (d) A close-up of the active site with a substrate (mostly green) bound. Two of the active-site residues, Ser195 and His57 (both red), are partly visible. Ser195 attacks the carbonyl group of the substrate (the oxygen is purple); the developing negative charge on the oxygen is stabilized by the oxyanion hole (amide nitrogens in orange), as explained in Fig ure 6–21. In the substrate, the aromatic amino acid side chain and the amide nitrogen of the peptide bond to be cleaved (protruding toward the viewer and projecting the path of the rest of the substrate polypeptide chain) are in blue.
The first evidence for a covalent acyl-enzymeinter mediate came from a classic application of pre–steady state kinetics. In addition to its action on polypeptides, chymotrypsin also catalyzes the hydrolysis of small esters and amides. These reactions are much slower than hydrolysis of peptides because less binding energy is available with smaller substrates, and they are therefore easier to study. Investigations by B. S. Hartley and B. A. Kilby in 1954 found that chymotrypsin hydrolysis of the ester p-nitro phenylacetate, as measured by release of p-nitrophenol, proceeded with a rapid burst before leveling off to a slower rate (Fig. 6–19). By extrapolating back to zero time, they concluded that the burst phase corresponded to just under one molecule of p-nitrophenol released for every enzyme molecule present. Hartley and Kilby suggested that this reflected a rapid acylation of all the enzyme molecules (with release of p-nitrophenol), with the rate for subsequent turnover of the enzyme limited by a slow deacylation step. Similar results have since been obtained with many other enzymes. The observation of a burst phase pro vides yet another example of the use of kinetics to break down a reaction into its constituent steps.
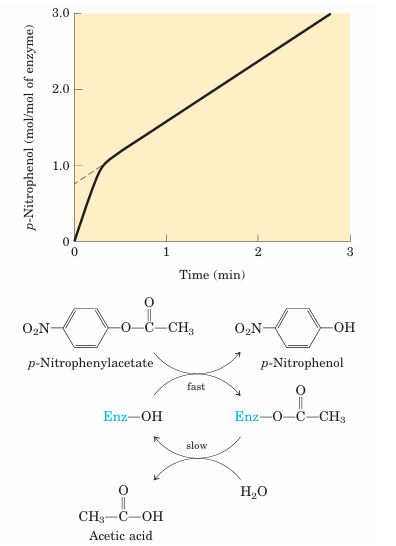
FIGURE 6–19 Pre–steady state kinetic evidence for an acyl-enzyme intermediate. The hydrolysis of p-nitro phenylacetate by chymotrypsin is measured by release of p-nitrophenol (a colored product). Initially, the reaction releases a rapid burst of p-nitrophenol nearly stoichio metric with the amount of enzyme present. This reflects the fast acylation phase of the reaction. The subsequent rate is slower, because enzyme turnover is limited by the rate of the slower deacylation phase.
Additional features of the chymotrypsin mechanism have been elucidated by analyzing the dependence of the reaction on pH. The rate of chymotrypsin-catalyzed cleavage generally exhibits a bell-shaped pH-rate pro file (Fig. 6–20). The rates plotted in Figure 6–20a are obtained at low (subsaturating) substrate concentrations and therefore represent kcat/Km. The plot can be dissected further by first obtaining the maximum rates at each pH, and then plotting kcat alone versus pH (Fig. 6–20b); after obtaining the Km at each pH, researchers can then plot 1/Km (Fig. 6–20c). Kinetic and structural analyses have revealed that the change in kcat reflects the ionization state of His57. The decline in kcat at low pH results from protonation of His57 (so that it cannot extract a proton from Ser195 in step 1 of the reaction; see Fig. 6–21). This rate reduction illustrates the importance of general acid and general base catalysis in the mechanism for chymotrypsin. The changes in the 1/Km term reflect the ionization of the -amino group of Ile16 (at the amino-terminal end of one of chymotrypsin’s three polypeptide chains). This group forms a salt bridge to Asp194, stabilizing the active conformation of the enzyme. When this group loses its proton at high pH, the salt bridge is eliminated and a conformational change closes the hydrophobic pocket where the
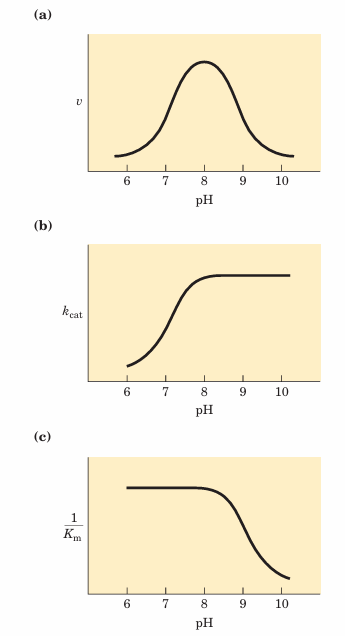
FIGURE 6–20 The pH dependence of chymotrypsin-catalyzed reactions. (a) The rates of chymotrypsin-mediated cleavage produce a bell-shaped pH-rate profile with an optimum at pH 8.0. The rate (v) being plotted is that at low substrate concentrations and thus reflects the term kcat/Km. The plot can be broken down to its components by using kinetic methods to determine the terms kcat and Km separately at each pH. When this is done (b and c), it becomes clear that the transition just above pH7 is due to changes in kcat, whereas the transition above pH 8.5 is due to changes in 1/Km. Kinetic and structural studies have shown that the transitions illustrated in (b) and (c) reflect the ionization states of the His57 side chain (when substrate is not bound) and the -amino group of Ile16 (at the amino terminus of the B chain), respectively. For optimal activity, His57 must be unprotonated and Ile16 must be protonated.
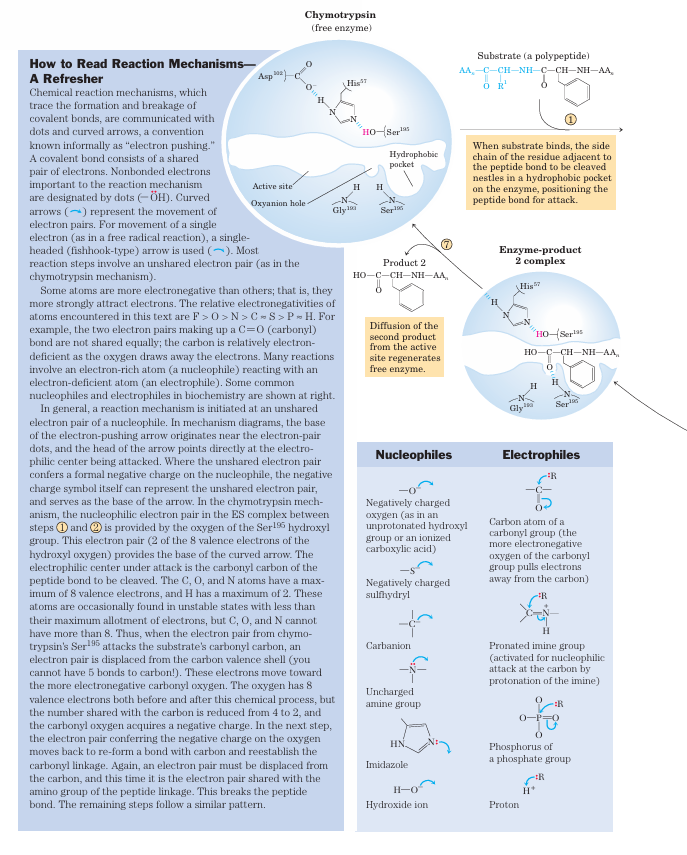
The tetrahedral intermediate in the chymotrypsin reaction pathway, and the second tetrahedral intermediate that forms later, are sometimes referred to as transition states, which can lead to confusion. An intermediate is any chemical species with a finite lifetime, “finite” being defined as longer than the time required for a molecular vibration (~10 13seconds). A transition state is simply the maximum-energy species formed on the reaction coordinate and does not have a finite lifetime. The tetrahedral intermediates formed in the chymotrypsin reaction closely resemble, both energetically and structurally, the transition states leading to their formation and breakdown. However, the intermediate represents a committed stage of completed bond formation, whereas the transition state is part of the process of reaction. In the case of chymotrypsin, given the close relationship between the intermediate and the actual transition state the distinction between them is routinely glossed over. Furthermore, the interaction of the negatively charged oxygen with the amide nitrogens in the oxyanion hole, often referred to as transition-state stabilization, also serves to stabilize the intermediate in this case. Not all intermediates are so short-lived that they resemble transition states. The chymotrypsin acyl-enzyme intermediate is much more stable and more readily detected and studied, and it is never confused with a transition state.
aromatic amino acid side chain of the substrate inserts (Fig. 6–18). Substrates can no longer bind properly, which is measured kinetically as an increase in Km. The nucleophile in the acylation phase is the oxygen of Ser195. (Proteases with a Ser residue that plays this role in reaction mechanisms are called serine pro teases.) The pKa of a Ser hydroxyl group is generally too high for the unprotonated form to be present in significant concentrations at physiological pH. However, in chymotrypsin, Ser195 is linked to His57 and Asp102 in a hydrogen-bonding network referred to as the catalytic triad. When a peptide substrate binds to chymotrypsin, a subtle change in conformation compresses the hydro gen bond between His57 and Asp102, resulting in a stronger interaction, called a low-barrier hydrogen bond. This enhanced interaction increases the pKa of His57 from ~7 (for free histidine) to>12, allowing the His residue to act as an enhanced general base that can remove the proton from the Ser195 hydroxyl group. De protonation prevents development of a very unstable positive charge on the Ser195 hydroxyl and makes the Ser side chain a stronger nucleophile. At later reaction stages, His57 also acts as a proton donor, protonating the amino group in the displaced portion of the substrate (the leaving group).
As the Ser195 oxygen attacks the carbonyl group of the substrate, a very short-lived tetrahedral intermedi ate is formed in which the carbonyl oxygen acquires a negative charge (Fig 6-21). This charge, forming within a pocket on the enzyme called the oxyanion hole, is stabilized by hydrogen bonds contributed by the amide groups of two peptide bonds in the chymotrypsin back bone. One of these hydrogen bonds (contributed by Gly193) is present only in this intermediate and in the transition states for its formation and breakdown; it re duces the energy required to reach these states. This is an example of the use of binding energy in catalysis.
 الاكثر قراءة في مواضيع عامة في الكيمياء الحياتية
الاكثر قراءة في مواضيع عامة في الكيمياء الحياتية
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


مواضيع ذات صلة
 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)