النبات

مواضيع عامة في علم النبات
الجذور - السيقان - الأوراق
النباتات الوعائية واللاوعائية
البذور (مغطاة البذور - عاريات البذور)
الطحالب
النباتات الطبية
الحيوان
مواضيع عامة في علم الحيوان
علم التشريح
التنوع الإحيائي
البايلوجيا الخلوية
الأحياء المجهرية
البكتيريا
الفطريات
الطفيليات
الفايروسات
علم الأمراض
الاورام
الامراض الوراثية
الامراض المناعية
الامراض المدارية
اضطرابات الدورة الدموية
مواضيع عامة في علم الامراض
الحشرات
التقانة الإحيائية
مواضيع عامة في التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحيوية والميكروبات
الفعاليات الحيوية
وراثة الاحياء المجهرية
تصنيف الاحياء المجهرية
الاحياء المجهرية في الطبيعة
أيض الاجهاد
التقنية الحيوية والبيئة
التقنية الحيوية والطب
التقنية الحيوية والزراعة
التقنية الحيوية والصناعة
التقنية الحيوية والطاقة
البحار والطحالب الصغيرة
عزل البروتين
هندسة الجينات
التقنية الحياتية النانوية
مفاهيم التقنية الحيوية النانوية
التراكيب النانوية والمجاهر المستخدمة في رؤيتها
تصنيع وتخليق المواد النانوية
تطبيقات التقنية النانوية والحيوية النانوية
الرقائق والمتحسسات الحيوية
المصفوفات المجهرية وحاسوب الدنا
اللقاحات
البيئة والتلوث
علم الأجنة
اعضاء التكاثر وتشكل الاعراس
الاخصاب
التشطر
العصيبة وتشكل الجسيدات
تشكل اللواحق الجنينية
تكون المعيدة وظهور الطبقات الجنينية
مقدمة لعلم الاجنة
الأحياء الجزيئي
مواضيع عامة في الاحياء الجزيئي
علم وظائف الأعضاء
الغدد
مواضيع عامة في الغدد
الغدد الصم و هرموناتها
الجسم تحت السريري
الغدة النخامية
الغدة الكظرية
الغدة التناسلية
الغدة الدرقية والجار الدرقية
الغدة البنكرياسية
الغدة الصنوبرية
مواضيع عامة في علم وظائف الاعضاء
الخلية الحيوانية
الجهاز العصبي
أعضاء الحس
الجهاز العضلي
السوائل الجسمية
الجهاز الدوري والليمف
الجهاز التنفسي
الجهاز الهضمي
الجهاز البولي
المضادات الميكروبية
مواضيع عامة في المضادات الميكروبية
مضادات البكتيريا
مضادات الفطريات
مضادات الطفيليات
مضادات الفايروسات
علم الخلية
الوراثة
الأحياء العامة
المناعة
التحليلات المرضية
الكيمياء الحيوية
مواضيع متنوعة أخرى
الانزيمات

Selection Can Be Detected by Measuring Sequence Variation
 المؤلف:
JOCELYN E. KREBS, ELLIOTT S. GOLDSTEIN and STEPHEN T. KILPATRICK
المؤلف:
JOCELYN E. KREBS, ELLIOTT S. GOLDSTEIN and STEPHEN T. KILPATRICK
 المصدر:
LEWIN’S GENES XII
المصدر:
LEWIN’S GENES XII
 الجزء والصفحة:
الجزء والصفحة:
 15-3-2021
15-3-2021
 2601
2601




+
-
20
Selection Can Be Detected by Measuring Sequence Variation
KEY CONCEPTS
-The ratio of nonsynonymous to synonymous substitutions in the evolutionary history of a gene is a measure of positive or negative selection.
-Low heterozygosity of a gene might indicate recent selective events.
-Comparing the rates of substitution among related species can indicate whether selection on the gene has occurred.
-Most functional genetic variation in the human species affects gene regulation and not variation in proteins.
Many methods have been used over the years for analyzing selection on DNA sequences. With the development of DNA sequencing techniques in the 1970s , the automation of sequencing in the 1990s, and the development of high-throughput sequencing in the 21st century, large numbers of partial or complete genome sequences are becoming available. Coupled with the polymerase chain reaction (PCR), which amplifies specific genomic regions, DNA sequence analysis has become a valuable tool in many applications, including the study of selection on genetic variants.
There is now an abundance of DNA sequence data from a wide range of organisms in various publicly available databases. Homologous gene sequences have been obtained from many species as well as from different individuals of the same species.
This allows for determination of genetic changes among species with common ancestry as compared to changes within a species. These comparisons have led to the observation that some species (e.g., D. melanogaster) have high levels of DNA sequence polymorphism among individuals, most likely as a result of neutral mutations and random genetic drift within populations. (Other species, such as humans, have moderate levels of polymorphism, and without further investigation, the relative roles of genetic drift and selection in keeping these levels low is not immediately clear.
This is one use for techniques to detect selection on sequences.) By conducting both interspecific and intraspecific DNA sequence analysis, the level of divergence due to species differences can be determined.
Some neutral mutations are synonymous mutations, but not all synonymous mutations are neutral. Although at first this might seem unlikely, the concentrations of individual tRNAs that specify a particular amino acid in a cell are not equal. Some cognate transfer RNAs (tRNAs) (different tRNAs that carry the same amino acid) are more abundant than others, and a specific codon might lack sufficient tRNAs, whereas a different codon for the same amino acid might have a sufficient number. In the case of a codon that requires a rare tRNA in that organism, ribosomal frameshifting or other alterations in translation may occur . It also might be that a particular codon is necessary to maintain mRNA structure. Alternatively, there might be a nonsynonymous mutation to an amino acid with the same general characteristics, with little or no effect on the folding and activity of the polypeptide. In either case neutral sequence changes have little effect on the organism. However, a nonsynonymous mutation might result in an amino acid with different properties, such as a changefrom a polar to a nonpolar amino acid, or from a hydrophobic amino acid to a hydrophilic one in a protein embedded in aphos pholipid bilayer. Such changes are likely to have functional effects that are deleterious to the role of the polypeptide and thus to the organism. Depending on the location of the amino acid in the polypeptide, such a change might cause only a slight disruption of protein folding and activity. Only in rare cases is an amino acid change advantageous; in this case the mutational change mightbecome subjected to positive selecti on and ultimately lead to fixation of this variant in the population.
One common approach for determining selection is to use codonbased sequence information to study the evolutionary history of a gene. Researchers can do this by counting the number of synonymous (Ks ) and nonsynonymous (Ka ) amino acid substitutions in orthologous genes and determining the Ka /Ks ratio. This ratio is indicative of the selective constraints on the gene. A Ka /Ks sratio of 1 is expected for those genes that evolve neutrally, with amino acid sequence changes being neither favored nor disfavored. In this case, the changes that occur do not usually affect the activity of the polypeptide, and this serves as a suitable control. A Ka /Ks ratio <1 is most commonly observed and indicates negative selection, where amino acid replacements are disfavored because they affect the activity of the polypeptide. Thus, there is selective pressure to retain the original functional amino acid at these sites in order to maintain proper protein function.
Positive selection is indicated when the Ka /Ks sratio is >1, but is rarely observed. This means that the amino acid changes are advantageous and might become fixed in the population. One example of this is the antigenic proteins of some pathogens, such as viral coat proteins, which are under strong selection pressure to evade the immune response of the host. A second example is some reproductive proteins that are under sexual selection (selection on traits found in one sex). As a third example, the Ka /Ks ratios for the peptide-binding regions of mammalian MHC genes, the products of which function in immunological self-recognition by displaying both “self” and “nonself” antigens, are typically in the range of 2 to 10, indicating strong selection for new variants. This is expected because these proteins represent the cellular
uniqueness of individual organisms.
The detection of a positive Ka /K s ratio might be rare in part because the average value must be greater than one over a length of sequence. If a single substitution in a gene is being positively selected, but flanking regions are under negative selection, the average ratio across the sequence might actually be negative. In contrast, the Ka /Ks ratios for histone genes are typically much less than one, suggesting strong negative selection on these genes.
Histones are DNA-binding proteins that make up the basic structureof chromatin and alterations to their structures are likely to result in deleterious effects on chromosome integrity and gene expression.
In addition to the difficulty of detecting strong selection on a single substitution variant when Ka /Ks is averaged over a stretch of DNA, mutational hotspots can also affect this measure. There have beenreports of unusually high ly mutable regions of some protein-coding genes that encode a high proportion of polar amino acids; such a bias might influence the interpretation of the Ka /Ks ratio because a higher point mutation rate might be incorrectly interpreted as a higher substitution rate. The lesson seems to be that although codon-based methods of detecting selection can be useful, their limitations must be taken into account.
Researchers can use intraspecific DNA sequence analysis to detect positive selection by comparing the nucleotide sequence between two alleles or two individuals of the same species.
Nucleotide sequences are expected to evolve neutrally at a rate proportional to the mutation rate; variation in this rate at specific nucleotides affects the heterozygosity of a population (the proportion of heterozygotes for a particular locus). If a variant sequence is favored, the variant will increase in frequency and eventually become fixed in the population, and the site will show a reduction in nucleotide heterozygosity. Closely linked neutral variants can also become fixed, a phenomenon termed genetic hitchhiking. These regions are characterized by having a lower level of DNA sequence polymorphism. (However, it is important to remember that reduced polymorphism can have other causes, such as negative selection or genetic drift.)
In practice it is more reliable to carry out both interspecific and intraspecific DNA sequence comparisons to detect deviations from neutral evolutionary expectations. By including sequence information from at least one closely related species, speciesspecific DNA polymorphisms can be distinguished from ancestral polymorphisms, and more accurate information regarding the link between the polymorphisms and between species differences can be obtained. With this combined analysis, the degree of nonsynonymous changes between species can be determined. If evolution is primarily neutral, the ratio of nonsynonymous to synonymous changes within species is expected to be the same as the ratio between species. An excess of nonsynonymous changes might be evidence for positive selection on these amino acids, whereas a lower ratio might indicate that negative selection is conserving sequences.
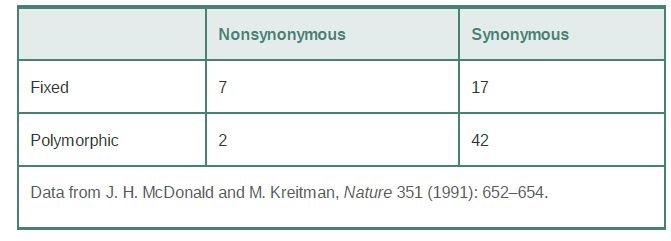
One example is the comparison of 12 sequences of the Adh gene in D. melanogaster to each other and to Adh sequences from Drosophila simulans and Drosophila yakuba, as shown in TABLE 1. A simple contingency chi-square test on these data shows that there are significantly more fixed nonsynonymous changes between species than similar polymorphisms in D. melanogaster. The high proportion of nonsynonymous differences among species suggests positive selection on Adh variants in these species, as does the lower proportion of such differences in one species, given that nonneutral variation would not be expected to persist for very long within a species.
TABLE 1 Nonsynonymous and synonymous variation in the Adh locus in Drosophila melanogaster (“polymorphic”) and between D. melanogaster, D. simulans, and D. yakuba (“fixed”).
Relative rate tests can also be used to detect the signature of selection. This involves (at a minimum) three related species: two that are closely related and one outgroup representative. The substitution rate is compared between the close relatives, and each is compared to the outgroup species to see if the substitution rates are similar. This removes the dependence of the analysis on time, as long as the phylogenetic relationships between the species are certain. If the rate of substitutions between related species compared to the rate between these and the outgroup species is different, this might be an indication of selection on the sequence.
For example, the protein lysozyme, which functions to digest bacterial cell walls and is a general antibiotic in many species, has evolved to be active at low pH in ruminating mammals, where it functions to digest dead bacteria in the gut. FIGURE 1 shows that the number of amino acid (i.e., nonsynonymous) substitutions for lysozyme in the cow/deer (ruminant) lineage is higher than that of the nonruminant pig outgroup.
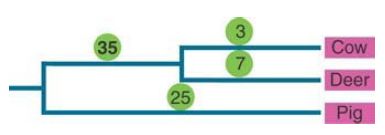
FIGURE 1. A higher number of nonsynonymous substitutions in lysozyme sequences in the cow/deer lineage as compared to the pig lineage is a result of adaptation of the protein for digestion in ruminant stomachs.
Data from: N. H. Barton, et al. 2007. Evolution. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. Original figure appeared in Gillespie J. H. 1994. The Causes of Molecular Evolution. Oxford University Press.
This method must take into account that some genes accumulate nucleotide or amino acid substitutions more rapidly (these are said to be fast-clock; see the next section A Constant Rate of Sequence Divergence Is a Molecular Clock) in some species than in others, possibly due to differences in metabolic rate, generation time, DNA replication time, or DNA repair efficiency. To deal with this difference, additional related species need to be examined in orderto iden tify and eliminate fast-clock effects. The reliability of this approach is improved if larger numbers of distantly related species are included. However, it is difficult to make accurate comparisons between taxonomic groups due to the inherent rate differences. As more work in this area has been done, corrections to adjust for differences in substitution rates have been developed.
Another method for detecting selection utilizes estimates of polymorphism at specific genetic loci. For example, sequence analysis of the Teosinte branched 1 (tb1) locus, an important gene in domesticated maize, has been used to characterize the nucleotide substitution rate in domesticated and wild maize (teosinte) varieties, with an estimate of 2.9 × 10-8 to 3.3 × 10-8 base substitutions per year. For a neutrally evolving gene, the ratio of a measure of nucleotide diversity (p) in domesticated maize to p in wild teosinte is about 0.75, but it is less than 0.1 in the tb1 region. The interpretation is that strong selection in domesticated maize has severely reduced variation for this gene.
As genome-wide data on nucleotide diversity become available, regions of low diversity can indicate recent selection. Millions of single nucleotide polymorphisms (SNPs) are being characterized in humans, nonhuman animals, and plants, as well as in other species. One approach that has been applied to the human genome is to look for an association between an allele’s frequency and its linkage disequilibrium with other genetic markers surrounding it. (Linkage disequilibrium is a measure of an association between an allele at one locus and an allele at a different locus.) When a new mutation occurs on one chromosome, it initially has high linkage disequilibrium with alleles at other polymorphic loci on the same chromosome. In a large population, a neutral allele is expected to rise to fixation slowly, so recombination and mutation will break up associations between loci and linkage disequilibrium will decrease. On the other hand, an allele under positive selection will rise to fixation more quickly and linkage disequilibrium will be maintained. By sampling SNPs across the genome, researchers can establish a general background level of linkage disequilibrium that accounts for local variations in rates of recombination, and any significantly higher measures of linkage disequilibrium can be detected. FIGURE 2 shows the slowly decreasing linkage disequilibrium (measured by the increasing fraction of recombinant chromosomes) with increasing chromosomal distance from a variant of the G6PD locus that confers resistance to malaria in African human populations. This pattern suggests that this allele has been under strong recent selection—carrying along with it linked alleles at other loci—and that recombination has not yet had time to break up these interlocus associations.
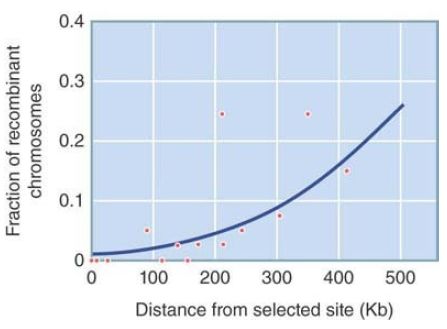
FIGURE 2. The fraction of recombinants between an allele of G6PD and alleles at nearby loci on a human chromosome remains low, suggesting that the allele has rapidly increased in frequency bypositi ve selection. The allele confers resistance to malaria.
Data from: E. T. Wang, et al. 2006. Proc Natl Acad Sci USA 103:135–140.
The availability of multiple complete human genome sequences an dthe ability to rapidly resequence specific regions of the genome in many individuals allows large-scale measurement of genetic variation in the human species. As described earlier, a lack of genetic variation in a stretch of DNA can indicate negative selection on that sequence, implying that the sequence is functional. If the analysis includes individuals from many populations, we can determine whether individual variations are unique, shared by other members of a specific population, or found globally. Surprisingly, such studies show that the majority of functional variations in the
human genome are not nonsynonymous changes in codingsequ ences, but are found in noncoding sequences such as introns or intergenic regions! In other words, protein variations account for only a small percentage of functional differences among humans.
Presumably, the large percentage of functional variation in noncoding regions reflects differences in regulatory regions . Also, most of these variations are found in most or all sampled populations and are not limited to one or a few populations. Clearly, despite many apparent differences among individual humans, there is genetic unity to the human species, and most of the differences are not with the proteins being produced in cells, but when and where they are being produced.
The 1000 Genomes Project began in 2008 with the initial goal of sequencing at least 1,000 individual anonymous human g enomes to assess comprehensive human genetic variation. During the first 2years of the project, sequencing progressed at a rate that was theequi valent of two genomes per day using reduced-cost, nextgeneration sequencing techniques. The sequence data are available in free-access public databases. By late 2015, more than 2,500 human genomes had been sequenced.
 الاكثر قراءة في مواضيع عامة في الاحياء الجزيئي
الاكثر قراءة في مواضيع عامة في الاحياء الجزيئي
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


مواضيع ذات صلة
 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)